5. Classheat: Large-scale profile analysis#
Churros provides a classheat function for clustering and visualizing large-scale epigenomic profiles.
This function takes regions of interest (e.g., specific protein binding sites) as input 1 and a folder of epigenomic signal files (either binary or continuous) as input 2.
In the binary mode,
classheatoutputs a binary matrix (output 1) representing the overlap of epigenomic markers at given genomic regions. The binary matrix is then formatted and sorted by the user-defined column (i.e., the filename of the selected marker) to generate the processed matrix (output 2) and plot the sorted heatmap (output 3). Subsequently,classheatutilizes PCA followed by k-means clustering (or other clustering methods) to produce the clustered matrix (output 4) and the clustered heatmap (output 5).In the continuous mode,
classheatcalculates the averaged read density of each epigenomic marker at given genomic regions (output 1). After logarithmic transformation, z-score normalization (optional method is 0-to-1 scaling), and sorting,classheatgenerates the remaining outputs in the same manner as in binary mode.
The main usages are:
churros_classheat mode region directory \
[-k kcluster] [-s sortname] [-l samplelabel] [-n normalize type] [-m cluster method]
The required parameters:
mode: either binary or continuous.
region: a BED format file for regions of interest (input 1). Only the first 3 columns are used.
directory: a directory containing the epigenomic signal files. The signal files can be either binary (e.g., peak files in BED format) or continuous (e.g., read coverage in bigwig format).
The optional parameters:
-k kcluster: number of clusters for clustered matrix and clustered heatmap. The default value is 3.
-s sortname: the filename of the selected marker in the directory above. This is used to for the processed matrix and sorted heatmap.
-l samplelabel: A .tsv table used to assign groups for each marker in the directory above. For example, it could look like this.
H3K27ac_ENCSR000EWR_rep1_peaks.narrowPeak |
H3K27ac |
GATA3_ENCSR000EWV_rep1_peaks.narrowPeak |
TFs |
H3K9me3_ENCSR000EWQ_rep3.mpbl.100.bw |
Histone |
Rad21_ENCSR000BTQ_rep1_peaks.narrowPeak |
TFs |
… |
… |
-n normalize type: Normalization methods for continuous data, could be zscore or scale0to1. Default: zscore.-m clustering method: minikmeans, kmeans, spectral, meanshift, dbscan, affinity
5.1. Example usage of binary mode#
churros_classheat -l samplelabel.tsv binary Rad21_ENCSR000BTQ_rep1_peaks.narrowPeak ./peakdir/
This command takes as input a file representing regions of interest (Rad21_ENCSR000BTQ_rep1_peaks.narrowPeak) and a directory (./peakdir/) containing multiple epigenomic signals.
We also assigned labels to the files in the ./peakdir/ directory.
Five output files are generated:
Output1_raw_matrix.tsv
Output2_sorted_matrix.tsv
Output3_sorted_heatmap.png
Output4_kmeans_matrix.tsv
Output5_kmeans_heatmap.png
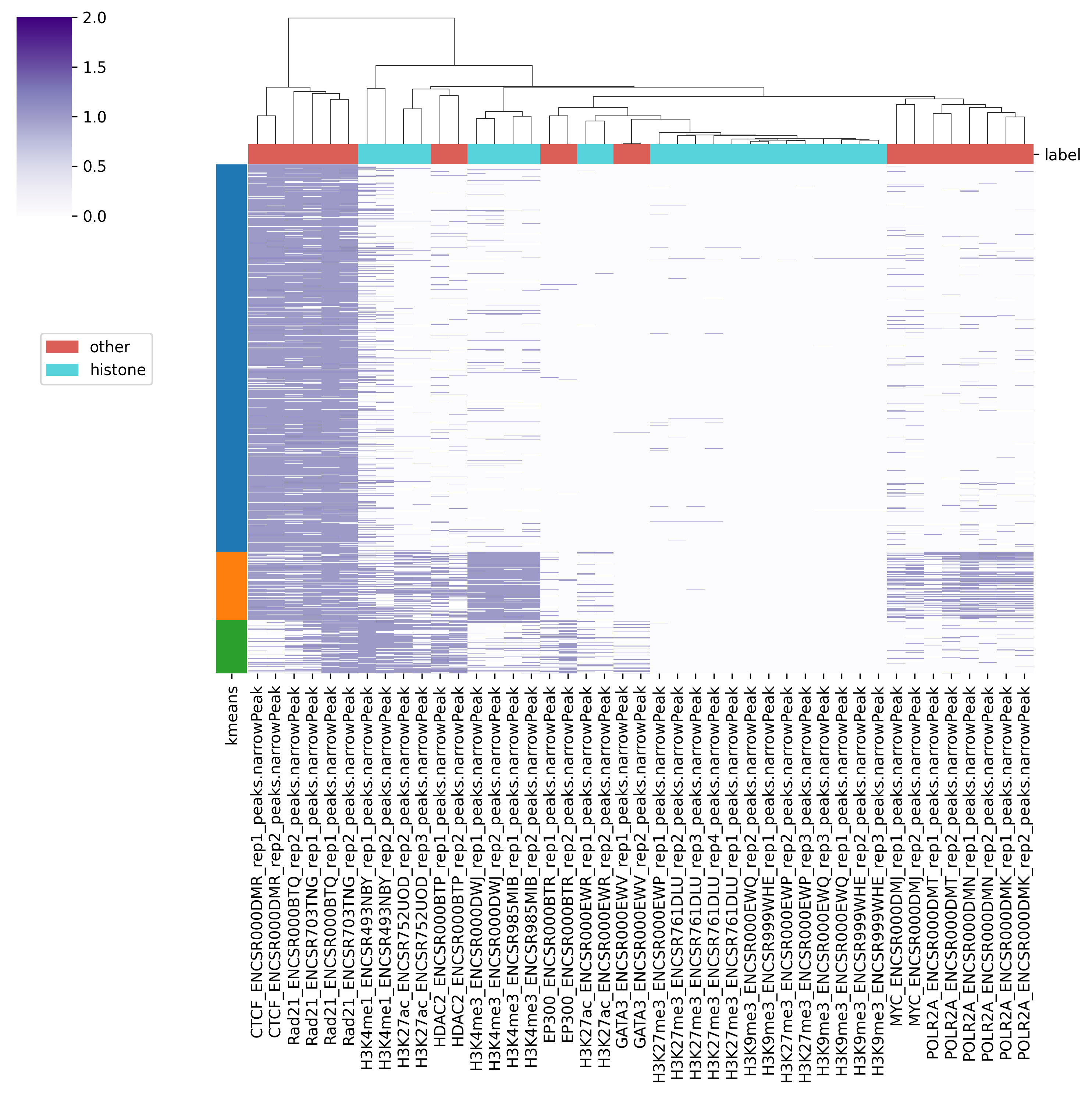
Fig. 5.1 Output5_kmeans_heatmap.png#
5.2. Example usage of continuous mode#
churros_classheat -l samplelabel.tsv -s GATA3_ENCSR000EWV_rep1.bw -k 3 -n zscore continuous Rad21_ENCSR000BTQ_rep1_peaks.narrowPeak ./bwdir/