3. Quick start (ChIP-seq, S. cerevisiae)#
This tutorial is for the analysis of S. cerevisiae. For human analysis, see Quickstart. The sample scripts are also available at Churros GitHub site.
Note
churros.sif). Please add apptainer exec churros.sif before each command below.apptainer exec churros.sif download_genomedata.sh3.1. Get data#
Here we use Scc1 and RPO21 ChIP-seq data from Jeppsson et al., Science Advance, 2022.
mkdir -p fastq
for id in SRR13065962 SRR13065963 SRR13065966 SRR13065967 SRR13065972 SRR13065973 SRR13065974 SRR13065975
do
fastq-dump --gzip $id -O fastq
done
download_genomedata.sh and build-index.sh.sacCer3 for genome build. See Reference Data Preparation for the detail of genome build.mkdir -p log
build=sacCer3
ncore=24
# download the genome
download_genomedata.sh -s $build Referencedata_$build/ 2>&1 | tee log/Referencedata_$build
# make Bowtie2 index
build-index.sh -p $ncore bowtie2 Referencedata_$build
3.2. Prepare sample list#
Churros takes two input files, samplelist.txt and samplepairlist.txt that describes the detail of samples to be analyzed.
3.2.1. samplelist.txt#
samplelist.txt is a tab-delimited file (TSV) that describes the sample labels and corresponding fastq files.
Multiple fastq files can be specified by separateing with ,.
Scc1_DMSO fastq/SRR13065962.fastq.gz
Input_Scc1_DMSO fastq/SRR13065963.fastq.gz
Scc1_thiolutin fastq/SRR13065966.fastq.gz
Input_Scc1_thiolutin fastq/SRR13065967.fastq.gz
RPO21_DMSO fastq/SRR13065972.fastq.gz
Input_RPO21_DMSO fastq/SRR13065973.fastq.gz
RPO21_thiolutin fastq/SRR13065974.fastq.gz
InputRPO21_thiolutin fastq/SRR13065975.fastq.gz
Note
See Prepare sample list for paired-end fastqs.
3.2.2. samplepairlist.txt#
samplepairlist.txt is a comma-delimited file (CSV) that describes the ChIP/Input pairs as follows:
ChIP-sample label
Input-sample label
prefix
peak mode
ChIP and input sample labels should be identical to those in samplelist.txt.
Input samples can be omitted if unavailable.
prefix is used for the output files.
peak mode is either [sharp|broad|sharp-nomodel|broad-nomodel]. This parameter is used for peak calling by MACS3.
Because MACS3 is not designed for small genomes such as yeast, the fragment length estimation sometimes fails.
Therefore here we specify sharp-nomodel that skips the model building.
Scc1_DMSO,Input_Scc1_DMSO,Scc1_DMSO,sharp-nomodel
Scc1_thiolutin,Input_Scc1_thiolutin,Scc1_thiolutin,sharp-nomodel
RPO21_DMSO,Input_RPO21_DMSO,RPO21_DMSO,sharp-nomodel
RPO21_thiolutin,InputRPO21_thiolutin,RPO21_thiolutin,sharp-nomodel
3.3. Running Churros#
churros command executes all steps from mapping reads to visualization.
Here we use --preset scer option to adjust the parameter setting to S. serevisiae.
churros --preset scer -p 12 samplelist.txt samplepairlist.txt sacCer3 Referencedata_sacCer3
-p 12 specifies the number of CPUs. sacCer3 is the UCSC genome build and Referencedata_sacCer3 is the directory generated by download_genomedata.sh and build-index.sh.
See Tutorial for the detail and output of churros_mapping, churros_callpeak and churros_compare.
3.4. churros_visualize: visualize read distributions by DROMPA+#
For a small genome (e.g., yeast), the sequencing depth is generally enough (> 10-fold). In such cases, the genome-wide ChIP/Input enrichment distribution is informative because the technical and biological bias in high throughput sequencing can be minimized.
churros_visualize has the option --enrich to visualize ChIP/Input enrichment in pdf format.
visualizes read distribution as pdf format.
churros_visualize samplepairlist.txt drompa+ \
sacCer3 Referencedata_sacCer3 --preset scer --enrich
--preset scer option changes the width and smoothing width for the small genome.
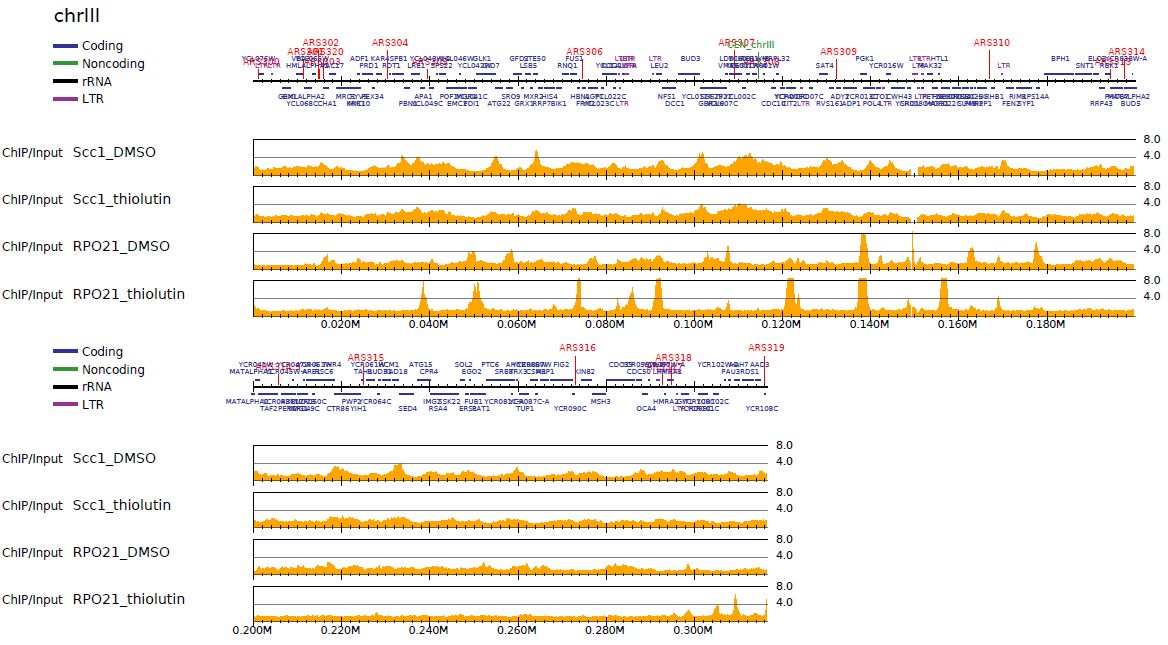
Fig. 3.1 ChIP/Input enrichment#
In addition, --logratio option makes the enrichment in log-scale.
churros_visualize samplepairlist.txt drompa+.logscale \
sacCer3 Referencedata_sacCer3 --preset scer --enrich --logratio
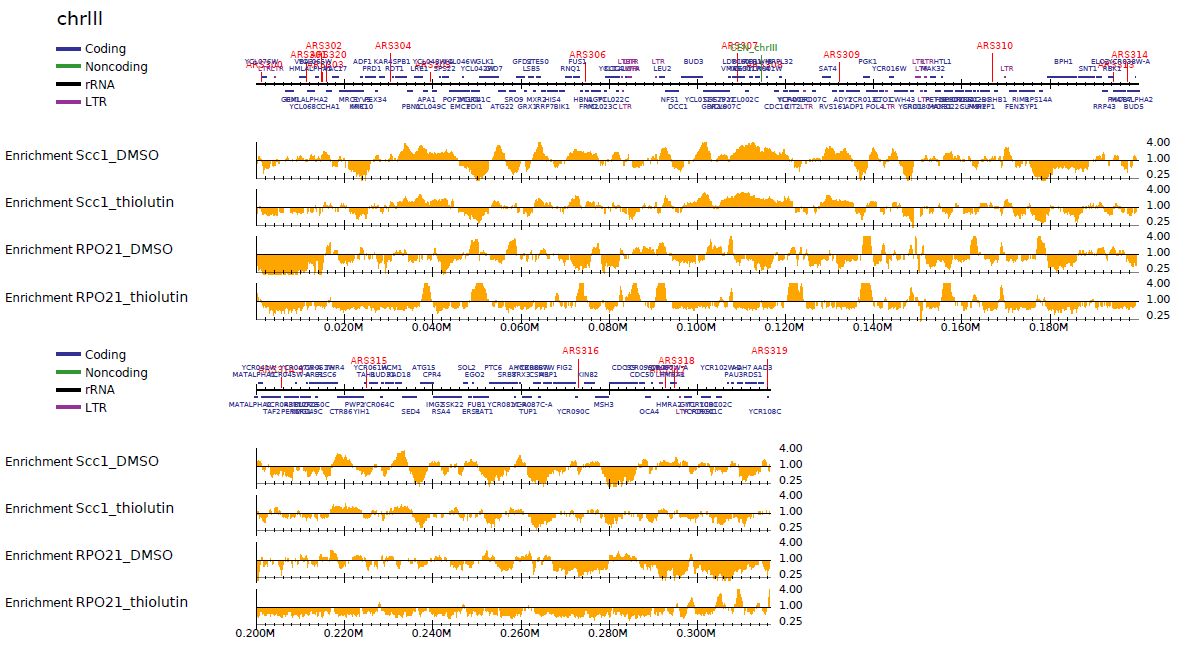
Fig. 3.2 ChIP/Input enrichment (log scale)#