4. Step-by-step analysis (ChIP-seq, human)#
Here we show the step-by-step ChIP-seq analysis using Churros. See also the sample scripts in the tutorial on GitHub.
Note
churros.sif). Please add apptainer exec churros.sif before each command below.apptainer exec churros.sif download_genomedata.sh4.1. Prepare sample list#
samplelist.txt is a tab-delimited file (TSV) that describes the sample labels and the path to the corresponding FASTQ files.
4.1.1. Single-end fastq files#
The sample labels and the path to the corresponding single-end FASTQ files are separated by tabs.
HepG2_H2A.Z fastq/SRR227639.fastq.gz
HepG2_H3K4me3 fastq/SRR227563.fastq.gz
HepG2_H3K27ac fastq/SRR227575.fastq.gz
HepG2_H3K27me3 fastq/SRR227598.fastq.gz
HepG2_H3K36me3 fastq/SRR227447.fastq.gz
HepG2_Control fastq/SRR227552.fastq.gz
Churros has a script gen_samplelist.sh to make the initial samplelist.txt.
gen_samplelist.sh fastq/ > samplelist.txt
4.1.2. Single-end, multiple fastq files#
If samples have multiple FASTQ files, specify the path to all FASTQ files connected with comma in the second column:
HepG2_H2A.Z fastq/SRR227639_rep1.fastq.gz,fastq/SRR227639_rep2.fastq.gz
HepG2_H3K4me3 fastq/SRR227563_rep1.fastq.gz,fastq/SRR227563_rep2.fastq.gz
HepG2_H3K27ac fastq/SRR227575_rep1.fastq.gz,fastq/SRR227575_rep2.fastq.gz
HepG2_H3K27me3 fastq/SRR227598_rep1.fastq.gz,fastq/SRR227598_rep2.fastq.gz
HepG2_H3K36me3 fastq/SRR227447_rep1.fastq.gz,fastq/SRR227447_rep2.fastq.gz
HepG2_Control fastq/SRR227552_rep1.fastq.gz,fastq/SRR227552_rep2.fastq.gz
4.1.3. Paired-end fastq files#
When using paired-end fastqs, use the second and the third columns to specify the R1 (F3) and R2 (F5) FASTQs:
HepG2_H2A.Z fastq/SRR227639_1.fastq.gz fastq/SRR227639_2.fastq.gz
HepG2_H3K4me3 fastq/SRR227563_1.fastq.gz fastq/SRR227563_2.fastq.gz
HepG2_H3K27ac fastq/SRR227575_1.fastq.gz fastq/SRR227575_2.fastq.gz
HepG2_H3K27me3 fastq/SRR227598_1.fastq.gz fastq/SRR227598_2.fastq.gz
HepG2_H3K36me3 fastq/SRR227447_1.fastq.gz fastq/SRR227447_2.fastq.gz
HepG2_Control fastq/SRR227552_1.fastq.gz fastq/SRR227552_2.fastq.gz
gen_samplelist.sh -p makes the samplelist.txt for paired-end samples.
Please do not separate paired-end fastqs with commas, as this will treat the reads as single-end.
gen_samplelist.sh -p fastq/ > samplelist.txt
4.1.4. Paired-end, multiple fastq files#
If samples have multiple FASTQ files, specify the path to all FASTQ files connected with comma in the second and the third columns for R1 and R2, respectively:
HepG2_H2A.Z fastq/SRR227639_rep1_1.fastq.gz,fastq/SRR227639_rep2_1.fastq.gz fastq/SRR227639_rep1_2.fastq.gz,fastq/SRR227639_rep2_2.fastq.gz
HepG2_H3K4me3 fastq/SRR227563_rep1_1.fastq.gz,fastq/SRR227563_rep2_1.fastq.gz fastq/SRR227563_rep1_2.fastq.gz,fastq/SRR227563_rep2_2.fastq.gz
HepG2_H3K27ac fastq/SRR227575_rep1_1.fastq.gz,fastq/SRR227575_rep2_1.fastq.gz fastq/SRR227575_rep1_2.fastq.gz,fastq/SRR227575_rep2_2.fastq.gz
HepG2_H3K27me3 fastq/SRR227598_rep1_1.fastq.gz,fastq/SRR227598_rep2_1.fastq.gz fastq/SRR227598_rep1_2.fastq.gz,fastq/SRR227598_rep2_2.fastq.gz
HepG2_H3K36me3 fastq/SRR227447_rep1_1.fastq.gz,fastq/SRR227447_rep2_1.fastq.gz fastq/SRR227447_rep1_2.fastq.gz,fastq/SRR227447_rep2_2.fastq.gz
HepG2_Control fastq/SRR227552_rep1_1.fastq.gz,fastq/SRR227552_rep2_1.fastq.gz fastq/SRR227552_rep1_2.fastq.gz,fastq/SRR227552_rep2_2.fastq.gz
4.1.5. Sample list with BAM files#
Starting with version 1.3.0, Churros can allow BAM files as input in samplelist.txt instead of FASTQ files. Just replace the FASTQ entries with BAM files like this:
HepG2_H2A.Z bam/HepG2_H2A.Z.sort.bam
HepG2_H3K4me3 bam/HepG2_H3K4me3.sort.bam
HepG2_H3K27ac bam/HepG2_H3K27ac.sort.bam
HepG2_H3K27me3 bam/HepG2_H3K27me3.sort.bam
HepG2_H3K36me3 bam/HepG2_H3K36me3.sort.bam
HepG2_Control bam/HepG2_Control.sort.bam
SAM (.sam), BAM (.bam) and CRAM (.cram) formats are acceptable. For paired-end map files, specify the --pair option to churros.
Note
Sample lists that contain both BAM and FASTQ files will not be accepted.
BAM files are only accepted in normal mode. The spike-in mode (
--spikein) does not allow BAM files as input.
4.2. Prepare sample pair list#
samplepairlist.txt is a comma-delimited file (CSV) that describes the ChIP/Input pairs as follows:
ChIP-sample label
Input-sample label
prefix
peak mode
HepG2_H2A.Z,HepG2_Control,HepG2_H2A.Z,sharp
HepG2_H3K4me3,HepG2_Control,HepG2_H3K4me3,sharp
HepG2_H3K27ac,HepG2_Control,HepG2_H3K27ac,sharp
HepG2_H3K27me3,HepG2_Control,HepG2_H3K27me3,broad
HepG2_H3K36me3,HepG2_Control,HepG2_H3K36me3,broad
ChIP and input sample labels should be identical to those in samplelist.txt.
prefix is used for the output files.
peak mode is either [sharp|broad|sharp-nomodel|broad-nomodel]. This parameter is used for peak calling by MACS3.
Input samples can be omitted if unavailable.
HepG2_H2A.Z,,HepG2_H2A.Z,sharp
HepG2_H3K4me3,,HepG2_H3K4me3,sharp
HepG2_H3K27ac,,HepG2_H3K27ac,sharp
HepG2_H3K27me3,,HepG2_H3K27me3,broad
HepG2_H3K36me3,,HepG2_H3K36me3,broad
In addition, Churros also has a script gen_samplepairlist.sh to make the initial template of samplepairlist.txt.
gen_samplepairlist.sh samplelist.txt > samplepairlist.txt
4.3. churros_mapping: mapping reads#
churros_mapping takes FASTQ and maps reads to the genome specified by Bowtie2 by default.
The mapped reads are then quality-checked and converted to BigWig files.
build=hg38
Ddir=Referencedata_hg38
# mapping
$sing churros_mapping -p 12 exec samplelist.txt $build $Ddir
# output QC stats
$sing churros_mapping header samplelist.txt $build $Ddir > churros.QCstats.tsv
$sing churros_mapping stats samplelist.txt $build $Ddir >> churros.QCstats.tsv
- Output
bam/ … map files (BAM format in default) and index files
sspout/ … output of SSP (strand-shift profile) for quality check
bigWig/ … bigWig files (100 bp, 5 kbp and 100 kbp bins by default) with raw count (
RawCount) and total read normalization (TotalReadNormalized)log/ … log files
4.4. checkQC.py: Quality check of the input samples#
Quality check (QC) is an important step in verifying the reliability of the results obtained.
From verion 0.11.0. Churros provides a script checkQC.py to check the quality of all input samples.
build=hg38
checkQC.py Churros_result/$build/churros.QCstats.tsv samplepairlist.txt
If the samples do not meet the criteria, the script will output a warning message.
See the checkQC.py: check the quality of the input ChIP-seq samples page for the detailed criteria.
4.5. churros_callpeak: call peaks by MACS2#
churros_callpeak calls peaks of the samples specified in samplepairlist.txt using MACS3.
If input samples are omitted, peaks are called using ChIP samples only.
By default, the churros command does not include this step. Add the --callpeak option to include this step in churros.
Note
Starting with version 1.4.0, the option of churros_callpeak to specify the number of CPUs has been changed from -p to -t to add a new parameter -p (paired-end mode).
churros_callpeak -t 8 samplepairlist.txt hg38
- Output
macs/ … peak files called by MACS3. The log files are stored in *log.
samplepairlist.txtinmacs/directory includes the filename of peak files that is used inchurros_visualize.
4.6. churros_visualize: visualize read distributions#
churros_visualize visualizes the distribution of raw reads, ChIP/Input enrichment and ChIP/Input p-value in PDF format using DROMPAplus.
The pdf files and corresponding peak lists are generated in pdf/.
churros_visualize samplepairlist.txt drompa+ hg38 Referencedata_hg38
To specify binsize 5-kbp, supply -b 5000. -l 8000 means the line size for each page is 8-Mbp. -P "--scale_tag 100" indicates the scale of y-axis is 100.
churros_visualize -b 5000 -l 8000 -P "--scale_tag 100" samplepairlist.txt \
drompa+.bin5M hg38 Referencedata_hg38
4.6.1. Highlight peak regions#
churros_visualize can highlight peak regions if the peak file is specified in samplepairlist.txt.samplepairlist.txt for churros_visualize is <ChIP-sample>,<Input-sample>,<prefix>,<peakfile>).churros_callpeak generated Churros_result/$build/macs/samplepairlist.txt that includes the peak files, churros_visualize highlights the peak regions by the command below:samplepairlist=Churros_result/hg38/macs/samplepairlist.txt
churros_visualize $samplepairlist drompa+.macspeak hg38 Referencedata_hg38
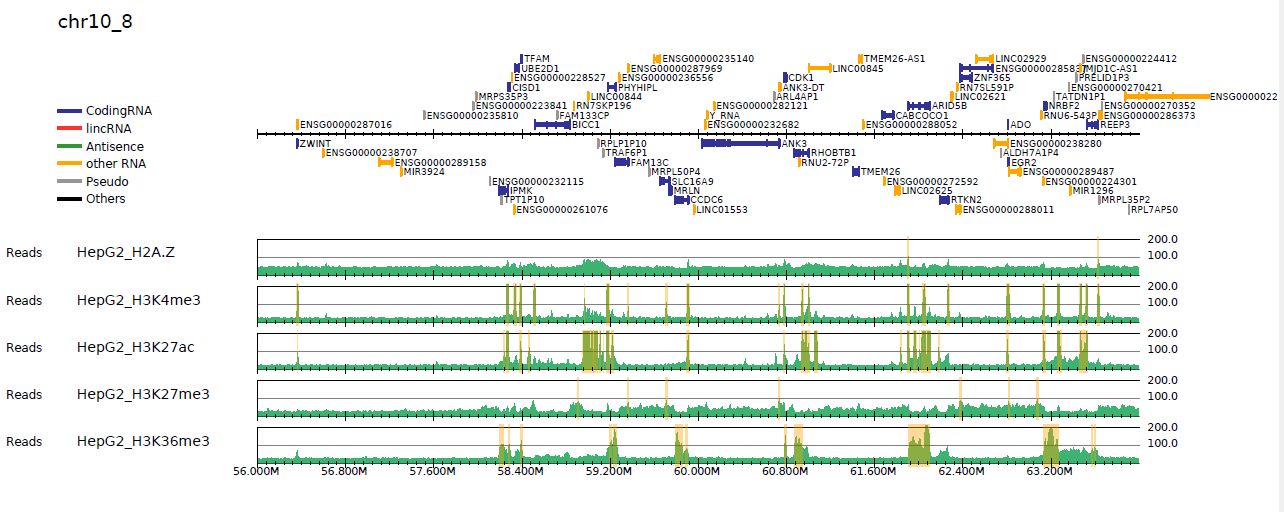
Fig. 4.1 Read distribution with peak highlighting#
4.6.2. Visualize p-value distribution#
Supply --pvalue option to visualize -log10(p) distribution of ChIP/input enrichment, which is recommended by ROADMAP project to distinguish the signal from the noise.
churros_visualize --pvalue -b 5000 -l 8000 \
samplepairlist.txt drompa+.pval.bin5M hg38 Referencedata_hg38
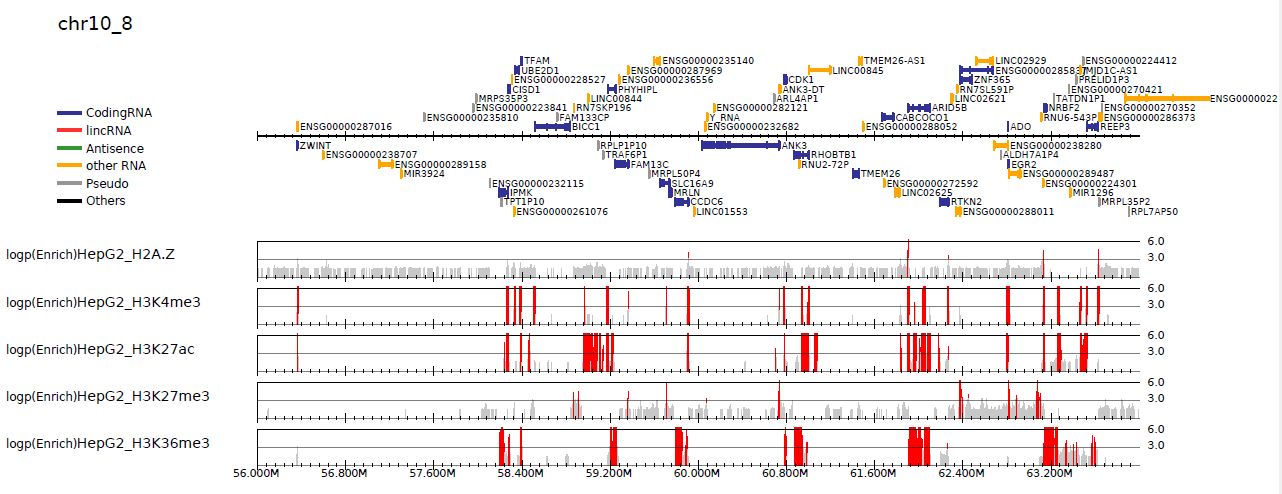
Fig. 4.2 -log10(p) distribution (ChIP/Input)#
4.6.3. Chromosome-wide view#
With the -G option, churros_visualize visualizes ChIP/Input enrichment for whole chromosomes with 100 kbp.
This option is useful for examining the euchromatin/heterochromatin pattern, for example.
churros_visualize -G samplepairlist.txt drompa+ hg38 Referencedata_hg38
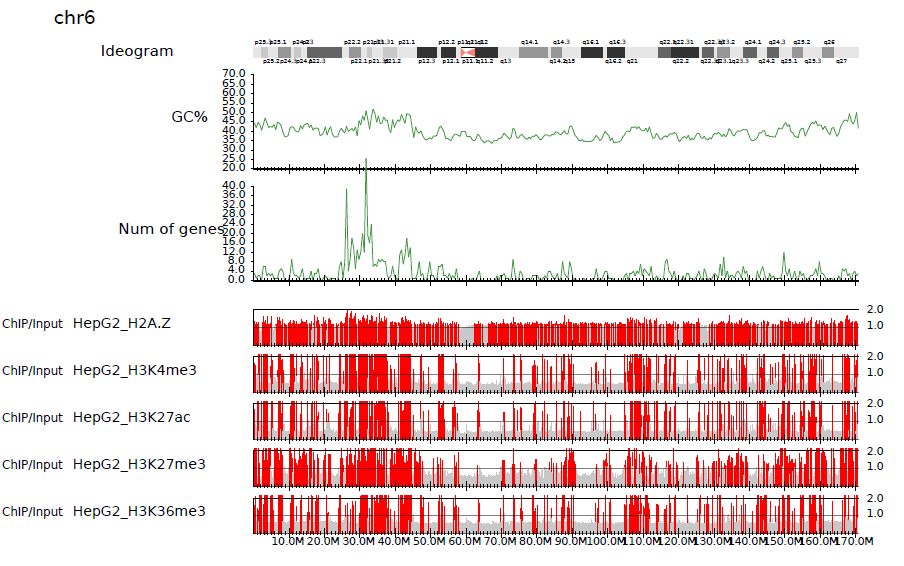
Fig. 4.3 Chromosome-wide distribution (ChIP/Input enrichment)#
4.6.4. Modify parameter sets for visualization manually#
churros_visualize also outputs a log file of pdf files generation
(e.g., `Churros_result/$build/log/pdf/drompa+.PCSHARP.100.log for Churros_result/$build/pdf/drompa+.PCSHARP.100.*.pdf).
This log file contains the command of DROMPA+ to make the pdf file at the top.
head -n1 Churros_result/$build/log/pdf/drompa+.PCSHARP.100.log
The output will look like this:
drompa+ PC_SHARP --ls 1000 -g Referencedata_hg38/gtf_chrUCSC/chr.gene.refFlat \
--gt Referencedata_hg38/genometable.txt --callpeak --showchr \
-i Churros_result/parse2wigdir+/HepG2_H2A.Z-bowtie2-hg38-raw-mpbl-GR.100.bw,Churros_result/parse2wigdir+/HepG2_Control-bowtie2-hg38-raw-mpbl-GR.100.bw,HepG2_H2A.Z, \
-i Churros_result/parse2wigdir+/HepG2_H3K4me3-bowtie2-hg38-raw-mpbl-GR.100.bw,Churros_result/parse2wigdir+/HepG2_Control-bowtie2-hg38-raw-mpbl-GR.100.bw,HepG2_H3K4me3, \
-i Churros_result/parse2wigdir+/HepG2_H3K27ac-bowtie2-hg38-raw-mpbl-GR.100.bw,Churros_result/parse2wigdir+/HepG2_Control-bowtie2-hg38-raw-mpbl-GR.100.bw,HepG2_H3K27ac, \
-i Churros_result/parse2wigdir+/HepG2_H3K27me3-bowtie2-hg38-raw-mpbl-GR.100.bw,Churros_result/parse2wigdir+/HepG2_Control-bowtie2-hg38-raw-mpbl-GR.100.bw,HepG2_H3K27me3, \
-i Churros_result/parse2wigdir+/HepG2_H3K36me3-bowtie2-hg38-raw-mpbl-GR.100.bw,Churros_result/parse2wigdir+/HepG2_Control-bowtie2-hg38-raw-mpbl-GR.100.bw,HepG2_H3K36me3, \
-o Churros_result/pdf/drompa+.PCSHARP.100 \
| tee -a Churros_result/pdf/drompa+.PCSHARP.100.log
Therefore, you can modify the resulting pdf files by directly modifying this command and -o option that specifies the output name.
For example, if you want to change the y-axis scale to 50, add --scale_tag 50 and execute:
drompa+ PC_SHARP --scale_tag 50 --ls 1000 (...) \
-o Churros_result/pdf/drompa+.PCSHARP.100.modified
See the manual for the detailed usage of DROMPAplus.
4.7. churros_compare: compare peaks between ChIP samples#
churros_compare outputs the heatmap of the correlation of peaks between ChIP samples.
The results are output to the comparsion/ directory.
Note
By default, the churros command does not include this step because the computation time becomes long when the number of samples is large. Add the --comparative option to include this step in churros.
If the number of peaks largely varies among samples, the comparison may become unfair.
Therefore, churros_compare also estimates peak overlap for top-ranked 2000 peaks.
churros_compare samplelist.txt samplepairlist.txt hg38
- The results include three types of comparisons.
bigwigCorrelation/… Spearman correlation of read distributions in 100 bp and 100 kbp bins from deepTools plotCorrelation. This score evaluates the similarity of the entire genome including non-peak regions. Therefore, the results may reflect the genome-wide features (e.g., GC bias and copy number variation) rather than peak overlap.Peak_BPlevel_overlap/… results of the base-pair level overlap of peaks (Jaccard index) using BEDtools jaccard. This score is good for broad histone modifications (e.g., H3K27me3 and H3K36me3).Peak_Number_overlap/… results of peak-number level comparison (Simpson index).PairwiseComparison/contains the results of all pairs (overlapped peak list and Venn diagram) and thePeakscontains the top-ranked peaks of samples. This score is good for comparing sharp peaks such as transcription factors.
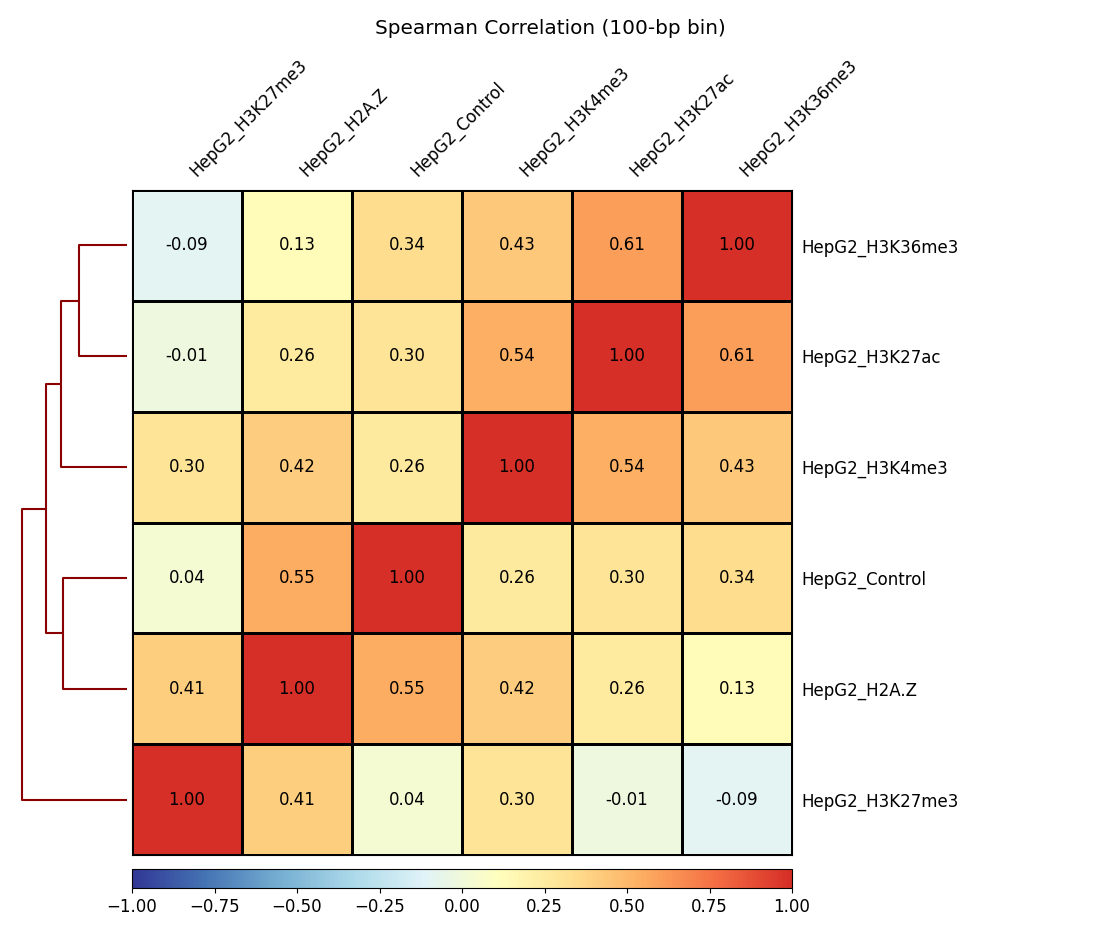
Fig. 4.4 bigwigCorrelation#
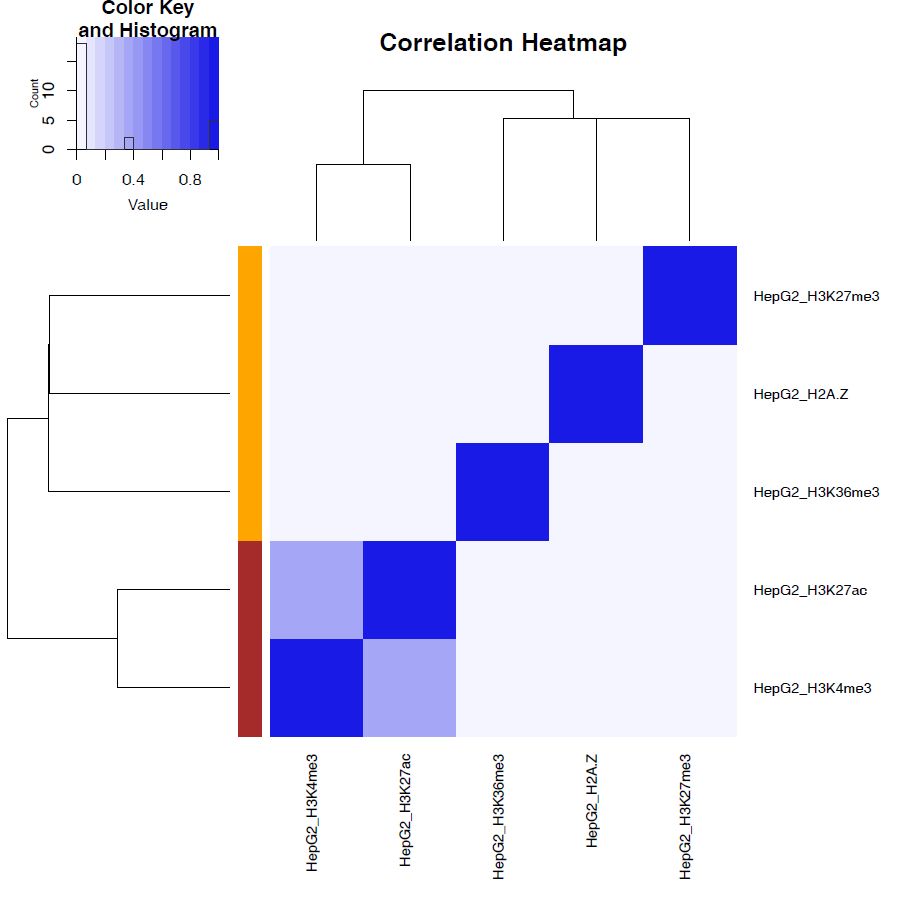
Fig. 4.5 Peak_Number_overlap#
4.8. churros_genPvalwig: generate P-value distribution as bedGraph#
churros_genPvalwig generates a -log10(P-value) distribution in bedGraph format. The P-value of upregulation and downregulation is output separately. The results are output in drompa+.pval/.
Note
By default, the churros command does not include this step. Add the --outputpvalue option to include this step in churros.
Ddir=Referencedata_hg38
gt=$Ddir/genometable.txt
churros_genPvalwig samplepairlist.txt drompa+.pval hg38 $gt