7. DNA methylation analysis#
This page describes how to analyze Bisulfite sequencing data for DNA methylation analysis with Churros. Churros includes Bismark to handle Bisulfite sequencing data. The sample scripts are also available at Churros GitHub site.
Note
churros.sif). Please add apptainer exec churros.sif before each command below.apptainer exec churros.sif download_genomedata.sh7.1. Get data#
Here we use two human RRBS data.
mkdir -p fastq
for id in SRR1609039 SRR1609040
do
fastq-dump --split-files --gzip $id -O fastq
done
download_genomedata.sh and build-index.sh.hg38 for genome build. See Appendix for the detail of genome build.mkdir -p log
build=hg38 # genome build
Ddir=Referencedata_$build # output directory
ncore=12 # number of CPUs
# download the genome
download_genomedata.sh -s $build $Ddir
# make Bismark index
build-index.sh -p $ncore bismark $Ddir
7.2. Running Bismark#
Bismark.sh command executes all steps of Bismark as follows:
bismark (mapping)
deduplicate_bismark
bismark_methylation_extractor
bismark2report
bismark2summary
In addition, Bismark.sh executes MultiQC to make a summary of quality statistics.
Supply -m option to specify the mode of Bisulfite sequencing ([directional|non_directional|pbat|rrbs]).
Because here we use a RRBS sample, -m rrbs option is supplied.
index=Referencedata_hg38/bismark-indexes_genome
ncore=24
Bismark.sh -p $ncore -m rrbs $index fastq/SRR1609039.fastq.gz
Bismark.sh -p $ncore -m rrbs $index fastq/SRR1609040.fastq.gz
The results are output in Bismarkdir/. If you want to specify the name of the output directory, use -d option.
- Output
*_bismark_bt2.bam … Map file by
bismark(BAM format)[CpG|CHG|CHH]_context_*_bismark_bt2.txt.gz … Output of
bismark_methylation_extractor. Context-dependent (CpG/CHG/CHH) methylation.*_bismark_bt2.bedGraph.gz … Bedgraph-format methylation information
*_bismark_bt2.bismark.cov.gz … Coverage file including counts methylated and unmethylated residues
*_bismark_bt2_*_report.html … Output of
bismark2report. Reports of Bismark alignment, deduplication and methylation extraction (splitting). Examplebismark_summary_report.html … Output of
bismark2summary. Summary of multiple Bismark data. Examplemultiqc_report.html … Output of MultiQC
See Bismark User Guide for more detail.
7.3. Methylkit for differential analysis#
You can then conduct differential analysis using methylKit. This is a sample R script.
library(methylKit)
files <- list("Bismarkdir/SRR1609039_trimmed_bismark_bt2.sorted.bam",
"Bismarkdir/SRR1609040_trimmed_bismark_bt2.sorted.bam"
)
sample.id <- list("SRR1609039", "SRR1609040")
myobj=processBismarkAln(location = files,
sample.id=sample.id,
assembly="hg38",
read.context="CpG",
mincov = 4,
treatment=c(0,1),
save.folder=getwd()
)
if (!dir.exists("methylKit")){
dir.create("methylKit")
}
pdf("methylKit/methyl_stats.pdf")
getMethylationStats(myobj[[1]],plot=TRUE,both.strands=FALSE)
dev.off()
pdf("methylKit/coverage_stats.pdf")
getCoverageStats(myobj[[2]],plot=TRUE,both.strands=FALSE)
dev.off()
meth=unite(myobj, destrand=FALSE)
pdf("methylKit/getCorrelation.pdf")
getCorrelation(meth,plot=TRUE)
dev.off()
pdf("methylKit/clusterSamples.pdf")
clusterSamples(meth, dist="correlation", method="ward", plot=TRUE)
dev.off()
pdf("methylKit/clusterSamples.pdf")
PCASamples(meth, screeplot=TRUE)
PCASamples(meth)
dev.off()
myDiff=calculateDiffMeth(meth, mc.cores=2)
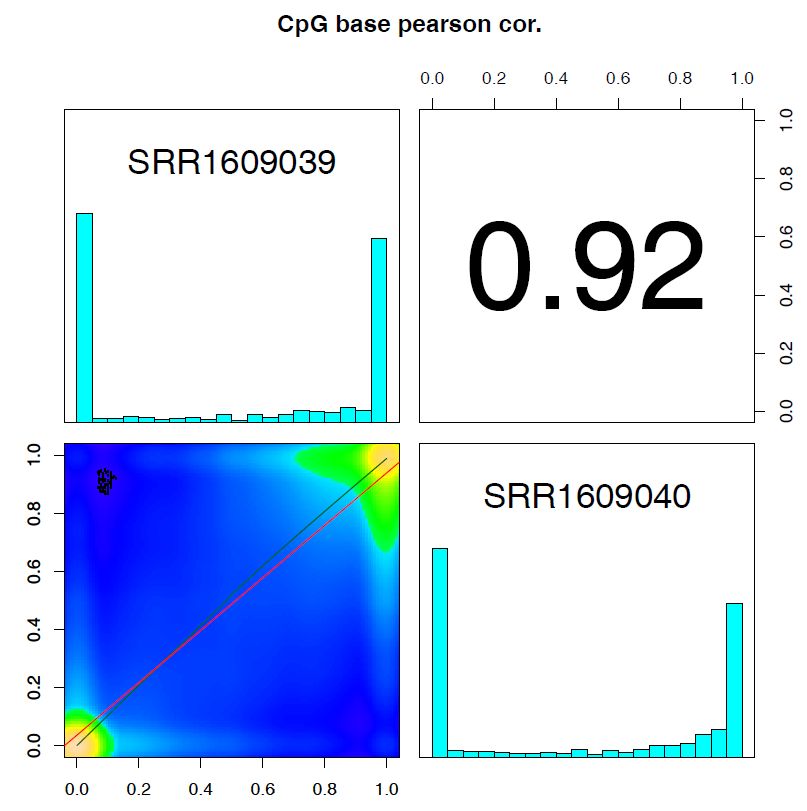
Fig. 7.1 getCorrelation.pdf#